The New Cost of Clinical Evidence

Over the last few months, I have been paying closer attention to how the cost of clinical evidence is shifting. Not only in dollars, but in the policy and operational layers that sit underneath every study we run in medtech. The landscape feels different than it did even a few years ago. Most of us already expect trials to be harder to run, harder to enroll, and harder to analyze. What is new this year is the set of forces around us that change how early companies plan, budget, and pace themselves.
Below are four areas that stood out to me: funding, diversity, geography, and coverage expectations. Together they are changing how device teams approach the path from prototype to data that matters.
1. SBIR reauthorization and the funding stack
Why this matters now: Early non-dilutive capital is tightening at the exact moment when more of the technical risk sits in the preclinical-to-FIH gap.
SBIR and STTR have always filled a very specific gap in medtech: non-dilutive funding for the early scientific and engineering work that happens long before private capital is ready to lean in. These programs give small teams enough runway to turn an idea into something that can enter a clinical workflow.
This year, that source of funding hit a pause. Authority expired when Congress did not complete reauthorization on time. NIH closed its SBIR and STTR announcements, and new grants won’t move until lawmakers agree on a path forward. With SBIR paused, early proof-of-concept work is shifting back onto equity, strategics, and founders themselves.
Among the US medtech companies that reached meaningful liquidity events this year, a visible subset had SBIR in their early funding stack. Monogram Orthopaedics, HeartFlow, and Beta Bionics all received SBIR or related government grants early in their development. It’s a pattern in companies with long technical arcs. NIH invested more than $1.3B into small businesses in FY2023, with a third flowing into medical technology.
Key Takeaway: If SBIR remains paused or is meaningfully altered, early-stage teams lose a tool that has historically supported this type of work.
2. FDA, diversity, and what sponsors need to plan for now
Why this matters now: Representation is moving from “best practice” to an operational requirement that affects timelines, budgets, and site strategy.
Another force shaping evidence is the expectation around who is represented in clinical trials. FDA estimates that roughly two-thirds of pivotal trials lack meaningful representation of key demographic groups. FDORA created a requirement for Diversity Action Plans in many later-stage studies. FDA issued guidance on how to structure those plans, then removed those documents from its site earlier this year after changes in federal DEI policy. The law itself hasn’t changed, and incoming HHS leadership has signaled that diversity requirements will continue.
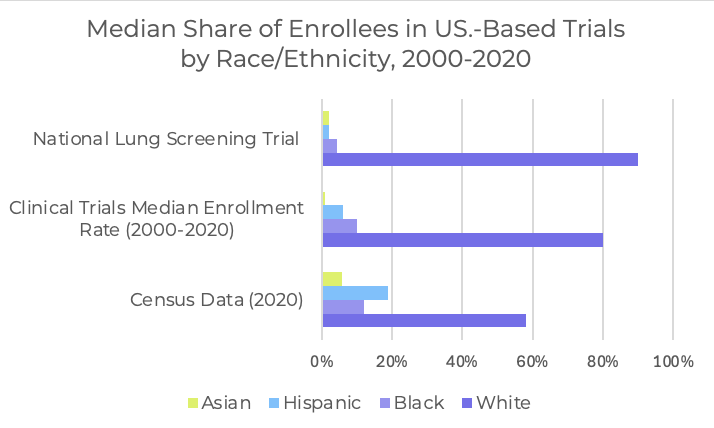
For device teams, this creates a mix of uncertainty and inevitability. The scientific need for broader representation is clear, and the operational impacts show up immediately. It shapes which sites you choose, how inclusion criteria are written, and how you resource enrollment. Balanced cohorts usually require more recruitment work, more community-focused outreach, and a wider geographic footprint. These steps add time and overhead, but they also build a more credible dataset and reduce the risk of later data requests.
Key Takeaway: If you wait for policy clarity, you lose time; representative enrollment needs to be planned now, even as specifics evolve.
Sources:
- Pew Research Center. (2025, November 3). Counting race: How the Census measures identity and what Americans think about it.
- Turner BE et al. Race/ethnicity reporting and representation in US clinical trials: a cohort study. The Lancet Regional Health – Americas. 2022
3. New geographies for device evidence
Why this matters now: The fastest paths to early data are increasingly outside the US, but only if quality and alignment with US regulatory expectations are maintained.
Another force shaping evidence is geography. Several regions have become attractive starting points for device studies, especially early-feasibility and first-in-human work.
Australia remains one of the strongest options. Blythe Karow's piece “Why Smart MedTech Money is Looking Down Under” has a lot of great insights on the advantage of pursuing clinical work in Australia. Faster first-patient-in, parallel contracting and ethics review, no full IDE, and the R&D tax incentive all contribute to predictable timelines and lower cost.
Australia timelines: ethics approval in 4–8 weeks; site activation in 8–12 weeks.
Cost differentials: up to 43.5% of eligible R&D spend returned; trials often 30–40% less than US equivalents.
Central Asia is also appearing more often in device study plans. Georgia and Uzbekistan offer quick activation, high-volume procedural centers, and lower per-patient costs. Sponsors report faster enrollment and strong investigator engagement in early device work.
Georgia/Uzbekistan timelines: activation in 6–10 weeks; enrollment 2–3× faster than comparable US sites.
Cost differentials: 40–70% lower than US or EU averages.
Key Takeaway: These regions offer speed and affordability, but the responsibility is ensuring that imaging, protocols, and data integrity meet US regulatory and payer expectations.
4. TCET and the new expectations around evidence design
Why this matters now: Coverage questions are moving upstream into early clinical design, and teams need to plan for economic and post-market outcomes earlier than ever.
As evidence becomes more global, it is also becoming more tightly connected to reimbursement. CMS’s Transitional Coverage for Emerging Technologies program is another shift worth paying attention to. TCET creates a path for certain breakthrough-designated devices to receive national Medicare coverage on a predictable timeline while new evidence is gathered. CMS explored a similar idea in its earlier MCIT proposal, which would have granted selected technologies four years of temporary coverage after FDA authorization. MCIT never took effect, but it helped shape the thinking behind TCET. Think of devices that aim to change referral pathways or reduce inpatient days. TCET is built for exactly that category.

CMS expects to accept only three to five devices per year and finalize coverage decisions within six months of FDA authorization. That is significantly faster than historical coverage patterns, where Medicare decisions often lagged FDA authorization by years.
Key Takeaway: Even if a company never applies for TCET, the program signals where payer expectations are heading: closer to early clinical design.
Sources:
- Sexton ZA et al. Time from authorization by the US Food and Drug Administration to Medicare coverage for novel technologies. JAMA Health Forum. 2023
- Centers for Medicare & Medicaid Services (CMS).Transitional Coverage for Emerging Technologies (TCET) Notice. CMS TCET Fact Sheet and Federal Register materials.
What I’m Watching Next
- How the UK implements its new pathway for accepting certain FDA decisions, and whether it meaningfully shortens time to market for US device companies.
- Whether the EU moves toward more flexible MDR reforms, especially for innovative or early-stage devices.
- Any progress on SBIR/STTR reauthorization, and what shape the program takes if it returns.
- The first set of devices CMS selects for TCET, which will signal the evidence patterns payers want to see earlier.
Closing
The cost of evidence keeps rising, but not just in dollars. It is rising in expectations, timelines, and the decisions we have to get right earlier. These shifts affect where we run, who we enroll, how we plan for coverage, and how we fund the messy middle between prototype and proof. The more we treat evidence as a core part of company building, rather than something we bolt on once the device is “ready,” the more room we give ourselves to move quickly without cutting corners.

